
More than half of all cancers have mutations in a gene called p53. The protein made from this gene is what’s called a tumor suppressor: When working properly, it guards against cancer development — in part, by detecting damaged DNA and alerting cells to repair it.
Cells without working p53 are unable to properly repair damaged DNA, leading to a buildup of mutations, including large chromosomal alterations. Because of its important role in maintaining DNA integrity, scientists long ago dubbed p53 the “guardian of the genome.”
But 30 years on from that christening, many questions remain about exactly how p53 guards the genome — and how its loss promotes cancer.
One hotly debated question has been whether the guardian role of p53 is important for preventing cancer. While tumors with p53 mutations show evidence of chromosomal alterations, research has shown that the normal p53 protein controls several other processes that might explain why its inactivation promotes cancer. For example, p53 promotes apoptosis, or programmed cell death, in cells that have developed precancerous features.
Another question relates to how the genetic instability arises following p53 loss. One longstanding assumption has been that p53 loss acts as a kind of “gateway to genetic chaos.” In other words, losing the tumor suppressor leads to random buildup of genetic mutations without much rhyme or reason. But a new study from researchers at the Sloan Kettering Institute (SKI) challenges that assumption and brings some insight into p53’s guardian role.
“Rather than promoting genetic chaos, what we see when cells lose p53 is an orderly progression of genetic changes that is actually quite predictable,” says Scott Lowe, Chair of the Cancer Biology and Genetics Program in SKI and the senior author on the study, which was published August 17, 2022, in the journal Nature. “That came as a complete surprise to us and suggests a new way to think about possibly treating cancer.”
Red and Green Tell a Story
Scientists have struggled to fully understand p53’s role in cancer, particularly its effects on the genome, in part because there are few good laboratory models that allow the study of p53 function at the earliest (benign) stages of tumor development, well before cells have acquired obvious cancerous properties.
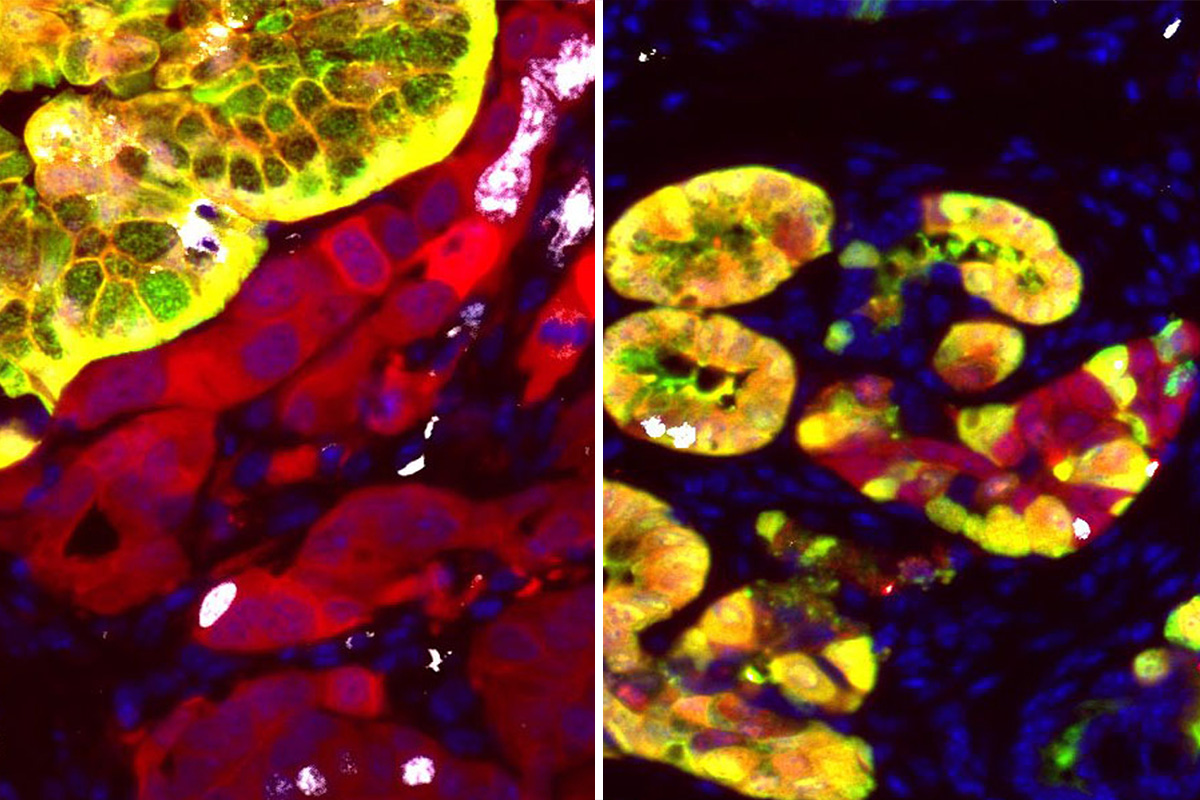
“The vast majority of cancer genomic studies are based on analyzing human tumors,” says Timour Baslan, the O’Neil Charitable Trust Fellow in the Lowe Lab and one of the paper’s lead authors. “The limited availability of patient tissue before and after tumor development means it has been impossible to gain a temporal picture of how p53 loss leads to cancer, starting from the earliest stages.”
To bring those early changes into view, Drs. Baslan and Lowe — along with former Lowe Lab members and cancer biologists Zhen Zhao and John P. Morris IV — produced a unique mouse model of pancreatic cancer in which p53 mutational status can be detected, irrespective of tumor development, thereby allowing measurements of genetic changes as incipient cancer cells transition from a benign to malignant state.
The model’s key feature is a set of fluorescent tags that record specific genetic events and can be detected with a microscope. One tag is red and records the presence of a mutated KRAS gene — known to be involved in promoting pancreatic cancer in both humans and mice. The other tag is green and records loss of p53. Cells with mutated KRAS but working p53 emit both red and green fluorescence, while cells that are missing p53 emit only red.
This visual trick allowed the scientists to identify specific populations of cells in the mouse that had lost p53 function but were still very far from being a full-fledged cancer. “It’s sort of like the first step when the wheels start to fall off the wagon,” Dr. Lowe says.
By collecting these specific cells and then performing single-cell DNA sequencing on them, the scientists were able to identify the genetic changes that occurred immediately following p53 loss and continuing after.
“The mouse really gave us the opportunity to look at a specific stage of cancer evolution, pull it out, and characterize it at a level that’s has never been done before,” Dr. Baslan says.
Order Within Chaos
To the scientists’ surprise, the changes they observed always seemed to happen in a consistent pattern. First, the cells lost particular regions of chromosomes — called deletions. Later on, genome doubling occurred, but only after a lot of deletions were accrued. Finally, following genome doubling, the cells continued to acquire further deletions but also uniquely gained additional copies of specific genes — called gains and amplifications.
“Since p53 mutations are often linked with genomic chaos, we were stunned to see there was a preferred order of events,” says Dr. Morris, now an assistant professor at the University of North Carolina at Chapel Hill.
Even though cells from early stages had lost p53, the researchers were able to show that they were not yet cancerous, but instead, required these changes to look and act like cancer cells. Together, these observations suggested to the researchers that p53 loss by itself is not sufficient to cause cancer; instead, cells lacking p53 must acquire additional genetic changes, in an orderly manner, to fully go rogue.
What’s true of the mouse also seems to be true of humans: The scientists could see evidence that the same sorts of deletions, doublings, and amplifications that occur in the mouse also occur in human pancreas tumors.
And it’s likely not just pancreatic cancer that follows this pattern. Since the team has started discussing their results with colleagues at Memorial Sloan Kettering Cancer Center (MSK), others have been finding similar changes in cancer types besides pancreatic cancer.
Treatment Implications
Knowing that there are “rules” to the genetic evolution of tumors suggests a different way of thinking about treating them, the scientists say.
Many existing cancer drugs target gene amplifications in tumors. But because these are acquired late in tumor evolution, not all cells in the tumor will have them. This means that drugs targeting these amplifications may kill off only certain cancer cells, leaving others unscathed.
A more effective approach to treating cancer might be to target the gene deletions that occur very early in cancer development, since these changes will be found in all, or nearly all, tumor cells. (Changes that occur early in tumor evolution are called truncal changes because they are found in the trunk of the tumor’s evolutionary tree.)
Targeting these deletions could be tricky, but Dr. Lowe says the possibility is there: “If it’s not genetic chaos, and there’s order and rules to cancer development, then you might ultimately be able to exploit those rules against the cancer itself,” he says.
An Important Anniversary
Fittingly, this new paper comes just after the 30th anniversary of the publication of the original Nature paper, by scientist David Lane, that named p53 the “guardian of the genome” in the first place. Since that time, scientists have developed a much deeper understanding of p53’s importance, with this latest paper bringing the multifaceted role of p53 into the sharpest focus yet.


